
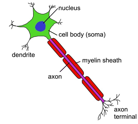
The basic unit of the nervous system is the nerve cell, also known as a neuron. These cells contain fibres called axons which send electrical impulses, and dendrites that branch out from the cell to receive these impulses.
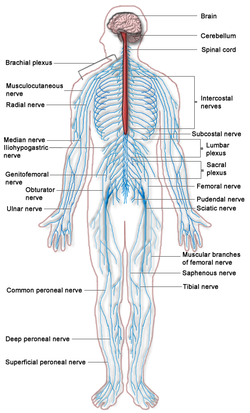
The nervous system has two distinct parts: the central nervous system (CNS) consisting of the brain and spinal cord, and the peripheral nervous system describing the nerves outside the brain and spinal cord.
Neuropathies describe disorders involving the peripheral nerves. If motor nerves (neurons transmit signals from the CNS to stimulate effector cells such as muscles) are damaged, muscles may weaken or become paralysed, and if sensory nerves (neurons that convey information, in the opposite direction, from tissues to the CNS) are damaged, sensation may be lost or abnormal sensations may be felt. The most common hereditary motor and sensory neuropathy is Charcot-Marie-Tooth disease.
Many diseases result from the degradation of myelin sheaths. These are the membranes surrounding neuronal axons which enables nerve impulses to travel quickly. Composed of fatty substances known as lipids, myelin can be likened to the insulation around an electrical wire, acting as an insulator to electrical signals. In demyelinating diseases myelin sheaths become degraded impeding the transport of impulses. The most common demyelinating disease is multiple sclerosis where the myelin is destroyed by the body’s own immune system possibly in response to some environmental trigger, and possibly in combination some level of genetic susceptibility. One of the most common inherited demyelination diseases is adrenoleukodystrophy.
A degeneration of neurons in the cerebellum and nerve tissue in the spinal cord, important for controlling muscle movement in the arms and legs can result in a progressive loss in coordination called ataxia, while another major group of diseases resulting from degeneration of neurons in the spine are hereditary spastic paraplegia.
Some neuropathies affect the autonomic system which regulates functions not under conscious control, such as heart rate or breathing. For instance the heart rate can increase for no apparent reason, or the kidneys suddenly fail to properly retain water. Several disorders associate with dysautonomia such as chronic fatigue system, the autoimmune multiple system atrophy and the inherited familial dysautonomia. One of the most severe dysautonomic diseases is known as Ondine’s Curse in which there is no autonomic control of breathing.
Degeneration of neurons or areas in the brain can cause characteristic clinical syndromes. Amyloidlateral sclerosis (also known as Motor neuron disease) causes paralysis, due to death of motor neurons in the motor cortex, brain stem and spinal cord while Huntington’s disease damages regions of the brain that control movement, emotion, and cognitive ability.
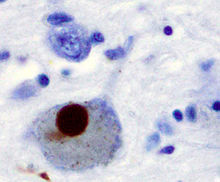
Parkinson’s disease results from loss of neurons in regions of the brain controlling movement and is characterised by unusual clumps of protein, known as Lewy bodies, building up in brain cells. Further types of young onset Parkinson’s disease include Parkin disease. A further disease characterised by these aggregations is Lewy body dementia, that shows degradation in cognition as well as motor control.
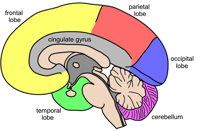
The most common neurodegenerative disease and the most common cause of dementia (Lat. de; away; mentis; mind) is Alzheimer’s disease. This disease also associates with the accumulation of clumps of amyloid protein, called plaques or tau protein as tangles. The aggregation of tau in different regions of the brain such as in the frontal and temporal lobes, causes frontotemporal dementia leading behavioural abnormalities rather than memory loss while progressive supranuclear palsy results from such aggregations in the midbrain.
Prion degenerative diseases result from an infectious agent that is neither bacterial, fungal, viral and in fact contains no genetic material at all but an altered protein.
Mental retardation is present in around 2 – 3% percent of the population; defined as cognitive ability that is markedly below average. Many environmental, genetic or multiple factors can cause mental retardation. A number of single-gene disorders include fragile X syndrome, neurofibromatosis, tuberous sclerosis, Noonan’s syndrome and as many as 25% of persons with mental retardation have a detectable chromosome abnormality such as Down syndrome, DiGeorge, Prader-Willi, Angelman and Williams syndromes.
LEWY BODY DEMENTIA
10-15% of diagnosed dementia
Lewy body dementia is characterised by symptoms similar to both Alzheimer’s disease and Parkinson’s disease but is in addition characterised by vivid and detailed hallucinations, fluctuating cognition and sleep disorders involving the REM period.
Like Alzheimer’s it is caused by accumulation of protein aggregates in the brain, but in this disease the protein aggregates are called Lewy bodies and consist of the protein alpha-synuclein.


Two well known writers whose work appears to have been affected by their encroaching Lewy body dementia are the Prussian philosopher Immanuel Kant and the British artist, poet and playwright, Mervyn Peake. Immanuel Kant showed clear symptoms of dementia, memory loss, repetitive behaviours (e.g. constantly buttoning and unbuttoning his clothes), fluctuations in his mental abilities and hallucinations. Mervyn Peake’s sketches during his illness portrayed the visual hallucinations he was experiencing while paranoid delusions become apparent in his later poetry.


Spanish author Miguel de Cervantes Saavedra may have incorporated the symptoms of Lewy body dementia into his writing. It is suspected that the behaviour of a patient he might have witnessed with the disorder was translated into the character Don Quixote in the novel of the same name. The eponymous hero suddenly becomes convinced that he is a knight errant. He dons an old suit of armour, renames himself Don Quixote de la Mancha, and sets off on fantastical adventures such as battling giants, which were in reality windmills. This novel ends with Don Quixote’s complete disillusionment and a melancholy return to sanity before his demise.
In a similar way, Charles Dickens may have based the character Ebenezer Scrooge on someone with Lewy body dementia.
In 2008, actress Estelle Getty, of the Golden Girls fame, died of Lewy body dementia. Cast members noted that years earlier, Getty had severe trouble remembering her lines during the filming of the show.

PRION DISEASES
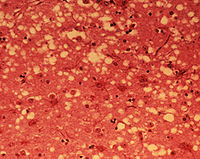
Prion diseases are rare infectious degenerative diseases of the brain characterised by massive neuronal death leaving open areas in the brain resembling a sponge – hence they are often known as transmissible spongiform encephalopathies.
These diseases are thought to be caused by a protein called PrPc that normally functions in cell signalling. This particular protein however, has the capacity to convert into an abnormal form called a prion (an acronym for proteinaceous infection). These can bind aggregate, and kill the cell in which it accumalates. These diseases can either be inherited, such as familial Creutzfeldt-Jakob disease (CJD), or acquired as in the variant CJD and kuru.
Mutations in different regions of the gene for PrPc can increase the likelihood of PrPc proteins converting to prions and causing a number of different autosomal dominant inherited prion diseases with overlapping symptoms such as familial CJD, Gerstmann-Sträussler-Scheinker syndrome and fatal familial insomnia that is characterised by the degeneration of an area which influences sleep.

The symptoms of CJD are a rapid progressive dementia and loss of coordinated movement, with death often occurring within six months of onset. George Balanchine, one of the 20th century’s greatest choreographers, died of CJD, which was diagnosed after his death. The first signs appeared in 1978 when he began losing his balance while dancing, followed soon after by deterioration in eyesight and hearing, until he became totally incapacitated. “I’m finished. I can’t hear, I walk like a drunken man” he said, shortly before dying a few years after developing the first symptoms at the age of 79.
Around one in every million people each year develops CJD, although this may be a low estimate; the disease is very difficult to diagnose as there is no definitive diagnosis without examining the brain.
Another prion disease affects the thalamus part of the brain controlling sleep. Fatal familial insomnia was first described an Italian country doctor, Ignazio Roiter, who married into a family suffering from this autosomal dominant condition. His wife’s aunts both died after developing confusion, paranoia and remaining sleepless, vegetating in a dream-like state for the last months of their lives. He was also able to trace the first appearance of this disease in the family over 250 years ago begining with the description of symptoms in a wealthy Venetian doctor who lived in the 1700s.

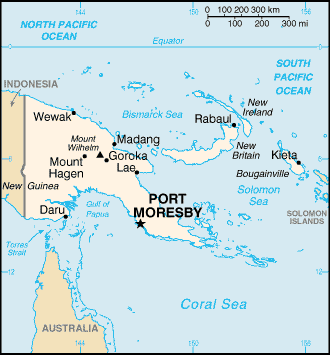
Only a small percentage of prion disease cases run in families with most prion diseases developing sporadically in the absence of any mutation or as a result of infection with abnormally formed prion protein. This was first noted in the Fore tribe of Papua New Guinea in the 1950s. During funeral rituals members of the tribe would eat the brains of dead relatives leading to the spread of a prion disease known as kuru, which means “trembling” in the language of the Fore. In a similar way, some cases of CJD in the U.S. have developed from the use of human growth hormone extracted from the pituitary glands of patients who died from CJD, though the known incidence of these cases is small and this procedure was discontinued in 1977.
It is possible that William Shakespeare might have provided the first account of CJD, in his character of Macbeth who experienced a rapid descent into madness and a decline in neurological and cognitive function accompanied with involuntary movements, hallucinations and insomnia. It is conceivable that he might have even had an understanding of the transmission of the disease by consumption of infected neuronal tissue: “…when the brains were out, the man would die. And there an end; but now they rise again. With twenty mortal murders on their crowns.” For Macbeth this might have been from the consumption of the brew given to him by the Weird Sisters, which contained various parts of humans and animals.

It is possible that William Shakespeare might have provided the first account of CJD, in his character of Macbeth who experienced a rapid descent into madness and a decline in neurological and cognitive function accompanied with involuntary movements, hallucinations and insomnia. It is conceivable that he might have even had an understanding of the transmission of the disease by consumption of infected neuronal tissue: “…when the brains were out, the man would die. And there an end; but now they rise again. With twenty mortal murders on their crowns.” For Macbeth this might have been from the consumption of the brew given to him by the Weird Sisters, which contained various parts of humans and animals.
CHARCOT-MARIE-TOOTH DISEASE
Different patterns of inheritance
1:2,500
Charcot-Marie-Tooth disease, named after the three doctors who discovered the gene in the early 20th century, usually result from damage of the membrane surrounding neuronal axons, known as the myelin sheath. Composed of fatty substances known as lipids, myelin can be likened to the insulation around an electrical wire, acting as an insulator to electrical signals, enabling nerve impulses to travel quickly. In demyelinating diseases myelin sheaths become degraded impeding the transport of impulses which would normally travel around 225 miles an hour. In this disease the loss of myelin leads to a slow progressive degeneration of muscles in the feet and lower legs which can spread to the hands and forearms, and generally presents in early adulthood. A number of mutations in different genes can produce this condition, the most common type affecting a gene producing a protein called peripheral myelin protein 22 which plays a role in the development and maintenance of myelin in the peripheral nerves.
Todd MacCulloch retired from playing basketball with the Philadelphia 76ers in 2003 after had been diagnosed with this condition which had been slowly eating away at his physical strength, starting with numbness and tingling in his feet and then spreading to his hands. He now works as a commentator.
Julie Newmar the American actress, dancer and singer, most famous role is that of Catwoman in the Batman television series also suffers and is now unable to walk without aid.

ARDENOLEUKODYSTROPHY

X-linked
1:10,000
Adrenoleukodystrophy is a metabolic disorder, in which the absence or malfunctioning of an enzyme results in the toxic accumulation of chemical substances damaging the nerves. The most common type is called X-linked ALD which results from the accumulation of high levels of Very Long Chain Fatty Acids (VLCFA) in the brain due to mutations in the ABCD1 gene that produces a protein necessary for the breakdown of VLCFA. This disease usually presents in early life when children that have previously developed normally, are quickly deprived of sight, hearing, speech and movement.
The film “Lorenzo’s Oil”, staring Nick Nolte and Susan Sarandon is based on the true story of Augusto and Michaela Odone, two parents in a relentless search for a cure for their son Lorenzo who was born in 1984 with the condition that had only been identified 10 years earlier. The Odones therefore set about learning everything possible about the disease and eventually came upon a treatment that seemed to prevent the disease from progressing any further in their child. For this, Augusto Odone received an honorary PhD. Known as Lorenzo’s Oil, the treatment is a mixture of two acids that prevents the build-up of VLCFA in the body. Outliving his expected life expectancy by two decades Lorenzo sadly died in 2008 at the age of 30 though the patent money from the oil continues to support an international research enterprise founded by the Odones called the Myelin Project.
Other mutations in this same ABCD1 gene result in a disorder known as Adrenomyeloneuropathy (AMN), which has an adult-onset, typically between the ages of 20 and 30. The Chilean musician Sebastian Santa Maria suffered from this. He was diagnosed with the disorder at the age of 34, while he was working on the recording of his first solo album “Latino”, and died 3 years later in 1996. His brother Julio had also suffered and died from AMN in the 1960s, at the age of 19.

HEREDITARY SPASTIC PARAPLEGIA
Many different forms of inheritance
1:30,000
Hereditary spastic paraplegia, describes a group of inherited neurological disorders in which only the legs gradually become weak. The age at onset and the degree of muscle weakness and spasticity may be extremely variable occurring as early as infancy or as late as old age. Initial symptoms typically include stiffness and relatively mild weakness of leg muscles, balance difficulties, unexplained trips and falls, and an unusually “clumsy” gait. As the disorder progresses, walking may become increasingly difficult although complete loss of the ability to walk is relatively rare, and lifespan is not affected. Mutations of many different genes may cause hereditary spastic paraplegia, and, in most cases, these mutations appear to be transmitted as an autosomal dominant trait, though some forms show either autosomal recessive or X-linked recessive inheritance. The symptoms appear to result from progressive degenerative changes of regions of the spinal cord that convey motor impulses from the brain to muscles involved in controlling certain voluntary movements.
ALZHEIMER'S DISEASE

1:1,400 (40-64yrs), 1:100 (65-69yrs), 1:25 (70-79), 1:6 (80+yrs)
Alzheimer’s disease is the most common type of dementia and occurs due to the degeneration of tissue in certain areas of the brain leading to a progressive deterioration in mental functioning. This particularly affects older people; about 1 in 5 over the age of 80 suffer from it.
While the causes of Alzheimer’s are still not precisely known, genetic factors are implicated in familial (usually autosomal dominantly inherited) and early-onset cases, which account for around 7% of all Alzheimer sufferers. The best-selling British author Terry Pratchett is suffering this form of early-onset Alzheimer’s disease. Even the late onset cases though, seem to have some genetic components with epidemiological studies suggesting that there is a higher chance of both twins suffering Alzheimer’s disease then there would be in the general population. Nevertheless, the only gene implicated so far is the APOE gene coding for a protein involved in the transport of lipids and in the metabolism of cholesterol, although it is still not properly understood how this leads to an increased risk.


One of the first effects of Alzheimer’s disease is the gradual loss of short-term memory and the ability to reason and concentrate. The British Prime Minister, Harold Wilson, first noticed these effects while still in office, becoming aware that his short term memory was no longer working as effectively, even though his long term memory appeared relatively unaffected. It was the realisation of this, that, in part, may have prompted his unexpected resignation in 1976. He may have been influenced by the experience of seeing the mental decline of his mother who died from Alzheimer’s disease.
After making his diagnosis of Alzheimer’s disease public in 1994, former President Reagan informed the nation of his condition with his typical optimism: “I now begin the journey that will lead me into the sunset of my life. I know that for America there will always be a bright dawn ahead. Thank you, my friends. May God always bless you.” But it was some years earlier, while he was in his second term of office, that some psychologists began to detect possible signs of the disorder in his conversation, speech and behaviour. At one press conference President Regan was asked about his plans for talks with the Russians on space weapons. He seemed confused by the question and was unable initially to find the words. Nancy Reagan whispered loudly, “Tell them we’re doing everything we can”. “We’re doing everything we can,” echoed the President. Remarkably, despite his growing dementia, he is remembered chiefly for his astute handling of the cold war, often against general advice. After leaving office as a hero, the disease went on to slowly ravage his mental capacity until he died of pneumonia on June 5, 2004.
PROGRESSIVE SUPRANEUCLEAR PALSY
Progressive supranuclear palsy shows only a low degre of inheritance – less than 1% of affected individuals have relatives with the disease. However some cases may result from mutations in the gene for tau leading to the tau protein aggregating in the midbrain. The midbrain is important in balance and eye-movements, so typical symptoms involve problems with control of balance, and an inability to control movements of the eye, particularly vertical movements. This results in the so-called “dirty-tie syndrome” because those affected cannot see that they are dropping food when they eat.
One well known sufferer was the British actor and comedian Dudley Moore. An accomplished pianist, he first noticed problems with the co-ordination of his finger movements while playing, later he would find it difficult to remember his lines while his developing slurred speech and loss of balance were misinterpreted by the public and the media as a sign of drunkenness. He was finally diagnosed when doctors found that he had the tell-tale restricted vertical eye movements which would later progress into more severe vision and balance problems and dementia. Before his death in 2002 he had set up the Dudley Moore Research Fund, raising awareness and funds for research in finding a cure for progressive supranuclear palsy.

PARKINSON'S DISEASE
1:500
The progressive movement disorder, Parkinson disease, is the second most common neurodegenerative disease after Alzheimer’s disease, affecting around 1 in 500 people, with symptoms usually appearing around the ages of 50 to 60.
The effects of Parkinson’s disease stem from the damage and loss of neurons in a part of the brain controlling muscle movement. These particular neurons make a chemical called dopamine which normally sends signals to coordinate movements. In a similar way to the neurodegenerative disorders previously discussed, aggregates of a protein, specifically alpha-synuclein in the case of Parkinson’s disease, are generally found in the affected brain.
For around 15 to 20 percent of patients there is some family history of the disease but the extent to which this is a result of shared genes rather than shared environmental risk is still uncertain. However, in the same way as Alzheimer’s disease, genetic factors appear to be more predominant when the disease begins earlier in life. Several genes are reported to underlie the young onset Parkinson’s disease, in which development of symptoms begin at 40 years or younger and tend to be inherited recessively. These include the previously mentioned the tau gene and the alpha-synuclein gene that also involved in Lewy body dementia. Mutations in another gene called parkin result in a disorder similar to Parkinson’s, called Parkin disease.
One prominent sufferer of young onset Parkinson’s is Michael J. Fox who first noticed symptoms at the young age of 29. He awoke one morning in 1990 with a persistent tremor of his left little finger; this then progressed to other areas of his body. He now focuses most of his energy on his Michael J. Fox Foundation for Parkinson’s Research which has raised much awareness and contributed many millions of dollars for research into the disorder. Michael was prescribed Levodopa, a dopamine substitute, to help control his symptoms allowing him to continue his acting career to 2000.


While Levodopa treatment for Parkinson’s is credited to the Swedish Nobel prize-winning scientist Arvid Carlsson, macuna seeds which contain levodopa are known to have been used in India to relieve the symptoms of palsy for at least 4,000 years.




Parkinson’s disease has played a major role in events of the last century by affecting a number of former dictators. It is likely that Parkinson’s disease was a key factor in Adolf Hitler’s downfall. He first began to show symptoms in 1934 with newsreels showing tremors in his hand and a shuffling walk. As with so many governments, his medical condition was kept secret and by the time of the Normandy landings, he had suffered from the disease for 10 years, and had in addition developed cognitive problems suggestive of dementia. Around a third of Parkinson’s suffers also develop dementia. However, Hitler would not be the last European fascist dictator to succumb to the effects of Parkinson’s disease. Francisco Franco was diagnosed with the disease in the 1960s and spent his last six years of rule in a highly weakened, often bed-ridden, state. Parkinson’s disease (or possibly motor neuron disease) has also been implicated in the death of the Chinese dictator, Mao Zedong in 1975. His successor Deng Xiaoping also suffered from the disease. Both men continued to rule for several years whilst being plagued with the symptoms.
PARKIN DISEASE
18% of early onset Parkinsons disease cases
Mutations in a gene called parkin result in a type of juvenile Parkinson disease called Parkin disease. Though sharing many clinical features, Parkin disease is pathologically very different to Parkinson’s disease. Though the same cells are affected in both cases, the brains of those with Parkin disease do not contain the typical protein deposits seen in Parkinson’s disease. While the majority of cases develop symptoms relatively early in life, most generally show a very slow progression. The parkin gene produces a protein is involved in attaching special molecules to defective proteins, tagging them for destruction – protein degradation is an important function because as proteins age, they can slowly become damaged and toxic to the cell.
Ozzy Osbourne was diagnosed with Parkin disease in 2005 claiming that he first noticed he had tremors in his 20s. “I’d always assumed it was the booze and stuff,” he explained, “Now I’ve found it all stems from the family. When I told my sister she said, ‘Not you as well? Mum had that and Auntie Elsie and your grandma.’ I’m like, ‘Thanks for f**king telling me’. Me walking around thinking I’ve got some drug paralysis.” He now takes medication to combat the involuntary tremors.

SPINOCEREBELLAR ATAXIA

Different types of inheritance
1:50,000
A progressive loss in coordination can result from a degeneration of neurons in the cerebellum and nerve tissue in the spinal cord, important for controlling muscle movement in the arms and legs. This can lead to the development of clumsy or awkward movements and unsteadiness, known as ataxia.
The most common recessively inherited ataxia is Friedrich’s ataxia resulting from mutations in a gene coding for the frataxin protein involved in mitochondria function. When this is disrupted nerve cells of the spinal cord are unable to inefficiently use cellular energy leading to their degeneration.
This disorder has a disproportionally high incidence among French Canadians which points to a founder effect. It seems that many inherited the mutant gene for FA from the married couple Jean Guyon and Mathurine Robin both of whom emigrated from Normandy in 1634.
The renowned British logician, Philip Edward Bertrand Jourdain, began to experience difficulties in walking as a young boy and by the time he became a student at Cambridge University he was crippled. While many sufferers do not survive past their twenties, Jourdain lived just short of his 40th birthday, dying in 1919.
While FA is recessively inherited, there are other forms of spinocerebellar ataxia which are dominantly inherited. These are caused by mutations in different genes encoding proteins found in nerve cells of the cerebellum and spinal cord. To date, there are 29 such genes identified, named SCA1-SCA29, most of which are affected, like FA, by triplet repeat mutations.
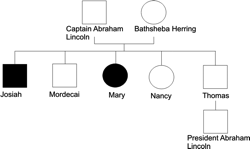
One of these genes (SCA5) is found in the family tree of President Abraham Lincoln. Either Captain Abraham Lincoln or Bathsheba Herring must have had the affected gene and passed it on to at least two of their five children who subsequently suffered from spinocerebellarataxia, and passed it further for 10 generations. President Lincoln would have had a 25 percent chance of inheriting the gene. As some of the other affected members of his family only developed symptoms as late as 68 years of age it could be possible that he himself had inherited the gene but symptoms had not yet manifested themselves by the time of his assassination at the age of 56.
HUNTINGTON'S DISEASE

Autosomal dominant
1:20,000
Huntington’s disease is caused by a triplet repeat mutation resulting in an altered Huntington protein which clumps together inside neuron cells leading to death. This particularly affects neurons in brain areas responsible for mental abilities and movement coordination with the progressive neuronal loss leading to the gradual development of abnormal movements and changes in cognition, behaviour, and personality. A persons’ ability to walk, think, talk and reason slowly diminish. The onset of symptoms is usually between the ages of 30 and 50 and it affects between 1 and 2 in 20,000 individuals.
It was in New York, in 1872, that George Huntington wrote a paper on the chorea emphasizing the hereditary nature of the disorder and providing the first scientific description of the disease. He was only 22 years old at the time! The family he studied were ancestors of a man by the name of Jeffrey Francis who emigrated from England, carrying the Huntington mutated gene, in 1634. This is an example of a ”founder effect” (described in more detail later) which also occurred in South Africa where several hundred people who developed Huntington’s disease were all descendents of a Dutch immigrant called Elsje Cloetens, who arrived there in 1652. Another population with a high incidence of the disease live near Lake Maracaibo in Venezuela and are all related to a Spanish sailor from Hamburg, Antonio Justo Doria, who lived during the 18th century and who travelled to Venezuela to buy dye for a German factory. It was the analysis of shared DNA sequences between these Venezuelan descendents that enabled scientists to ‘map’ the location of the Huntington gene to chromosome 4, by using a process known as Linkage analysis.


In America Huntington’s disease is often referred to as “Woody Guthrie’s disease” from the famous American folk singer who died from it. One day in the 1950s Woody Guthrie’s wife noticed her husband walking lopsidedly. This was soon followed by slurred speech and uncontrollable rages. Eventually, he lost all ability to talk, read, or walk and the only way he could communicate with his wife and children was by waving his arm at cards printed with the words ‘Yes’ and No’. In 1967 he died from the disease, which had previously killed his mother, from whom he had inherited it. This is because the disease is inherited in an autosomal dominant fashion and so a parent with the HD gene has a 50% chance of passing it on to each offspring. When a parent develops symptoms of HD their offspring have to decide whether or not to have themselves tested for the presence of the mutant gene. Should the results prove positive they would live with the certainty of developing symptoms of the disease before the age of 50. Insurance companies now legally have the right to know the outcome of any such test.
Sometimes known as Huntington’s Chorea, this disease has been reported since the 16th century and it was the early Renaissance physician Paracelsus who used the term “chorea” (the Greek word for dance) to describe the shaking and twitching that people with the disease went through. English colonists in America called the disease Saint Vitus Dance (St Vitus is the patron saint of dancers), and many of the “witches” at the infamous Salem Witch Trials of 1692 in Massachusetts, are now believed to have had HD; their choreic movements and odd behaviour were seen as possession by the devil.
Fragile X
X-linked dominant
1:in 4000
Fragile X syndrome is the most common inherited cause of intellectual disability varying in severity from mild to severe mental retardation with physical characteristics such as an elongated face and large or protruding ears.
It results from mutations to a gene on the X chromosome, required for normal neural development. Hence this X-inherited disease tends to more severely affect males than females. The type of mutation in this particular disorder is known as a trinucleotide repeat expansion where various numbers of a repeat (Fragile X syndrome this is the nucleotides CGG which can be repeated less than 50 times in unaffected and over 200 times in affected individualsd) disrupt the gene.


There are some discussions to what the twin brothers Dresie and Casie from Roger Ballen’s famous photograph (Transvaal, 1993: Courtesy of Roger Ballen and Hamilton’s Gallery, London) might suffer from. One suggestion is Fragile X syndrome mainly from their wildly protruding ears although they do not seem to show typical elongated faces and of course there is no cognitive (and genetic!) evidence as yet to support or rule out such an hypothesis.