
What is food to one man may be fierce poison to others.
Lucretius (99 BC – 55 BC)
There are many different diseases in which individuals lack a particular enzyme involved in converting various substances into other products, i.e. metabolism. Termed “inborn error of metabolism” by the British physician, Sir Archibald Garrod, in the early 1900s, the diseases highlight the fact that one gene produces one enzyme protein. In other words a mutation in one particular gene will only affect the one particular protein produced by that gene. While seemingly obvious now, this was a very astute observation at the time and helped pave the way for understanding the basis of genes and inheritance.
Metabolic diseases result from the loss of any one of a number of enzymes required for efficient metabolism of various substances such as carbohydrates (lactose intolerance, glycogen storage diseases, galactosemia), amino acids (phenylketonuria, maple syrup urine disease) and fats (Tay-Sachs disease). Disorders also result from the inability to break down some of the products of DNA (Lesch-Nyhan syndrome, gout) or defects in the organelles responsible for breaking down various substances (lysosomal storage disease, Zellweger syndrome). These diseases can either lead to the accumulation of toxic substances in body, or else a deficiency in specific biological compounds. The majority of these diseases show strict recessive inheritance, i.e. they are caused by a single gene and one defective copy of that gene must be inherited from each parent.
Lactose Intolerance
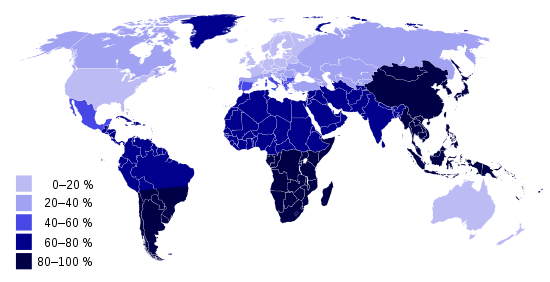
Mammals have evolved a devious trick to shorten the length of time an offspring is dependent on the mother. This involves a molecular mechanism in which the gene for lactase, needed for digesting milk, is turned off at the end of the weaning period. Lactase is an enzyme needed for the metabolism of the lactose sugar found in milk. So while young mammals are able to digest milk from the mother they will reach a stage shortly after weaning when the consumption of milk will incur abdominal cramps and diarrhoea. This also occurs in humans; a child’s bodily production of the lactase enzyme decreases dramatically after the first couple of years of life, resulting in lactose intolerance. Almost three quarters of the global population is lactose intolerant.
Most of the population of Europe and the US would still be milk intolerant if it were not for one or more individuals living around 4,000 BC in Sweden or the Middle East who acquired a particular mutation on chromosome 2. This has the effect of bypassing the normal shutdown in lactase production and so enabling the lifetime consumption of milk. This mutation proved to be a considerable evolutionary advantage following the first domestication of animals since milk then became a valuable source of nutriment. Most of Western Europeans have inherited this mutation and can safely consume milk, while many in African and Asian countries remain lactose intolerant.

After returning from the Beagle in 1836, Charles Darwin suffered for over 40 years from long bouts of vomiting and intestinal pain, among other undiagnosed ailments. These stomach problems would begin two to three hours after a meal, the time it takes for lactose to reach the large intestine. Furthermore, his family history seems to show a pattern of inheritance of this condition suggesting a genetic origin. Darwin only got better when, by chance, he stopped taking milk and cream.
Phenylketonuria

Phenylketonuria results when individuals inherit mutations in the phenylalanine hydroxylase gene which produces the enzyme needed to convert the amino acid phenylalanine into the amino acid tyrosine. The resulting higher levels of phenylalanine in the blood harm brain development and can lead to mental retardation if not treated. In addition, some of the excess phenylalanine is converted into a phenylketone called phenylpyruvic acid, which, when excreted in the sweat and the urine, gives a musty odour, hence the name for this condition, i.e. phenyketones in the urine.
Perhaps one of the first reports of phenylketonuria was by the Nobel prize-winning American author, Pearl Sydenstricker Buck, in her book “The Child Who Never Grew”. Here she described how her 3 year old daughter that she brought from China to the US in the 1920s started to become severely mentally retarded in early childhood. She wrote: “Although the child was beautiful, her mind was not developing. I remember when she was only 3 months old she lay in her basket on the sun deck of a ship. I had taken her there for the morning air. The people who promenaded on deck often stopped to look at her, and my pride grew as they spoke of her unusual beauty and of the intelligent look of her deep, blue eyes. I do not know at what moment the growth of her intelligence stopped.”
A decade later, the Norwegian physician, Ivar Asbjørn Følling, also noticed the same disease in children living near his hometown of Oslo. Seemingly healthily growing infants would, at some point, stop developing normally and drift into irreversible mental retardation. He wondered whether the disorder might have something to do with metabolism and so set about seeing if there might be any unbroken down products in the urine of the children. He found that the urine of all affected children contained phenylketone which is a derivative of the amino acid phenylalanine. Many years later Pearl Sydenstricker Buck also recalled the characteristic musty odour now associated with the disease. Følling’s discovery served two life-saving purposes: it allowed the diagnosis of this disorder, and also provided the cure, which simply involves restricting the diets of those affected children to only those foods containing low levels of phenylalanine. The photograph shows two siblings born with phenylketonuria; the untreated bother on the left and his sister who received the restricted diet.

Tay-Sachs Disease
Tay-Sachs disease occurs due to mutations disrupting the function of an enzyme needed to break down specific lipids which then accumulate in tissues, particularly neurons that use this type of lipid for membranes and synapses. Children with this disease become progressively retarded, with associated spasticity, paralysis, dementia, and blindness, ending with an early death, usually by 4 years of age. Autosomal recessive inherited, Tay-Sachs disease is most common in families of Eastern European Jewish origin, where as many as 1 in 45 are carriers of the mutant gene compared to the general incidence of 1 in 300. This disease has actually been described in the Ashkenazi Jewish population as early as the 15 century. This high incidence suggests a possible heterozygous advantage in this population. There is some evidence that carriers of the Tay-Sachs disease gene have an increased resistance to Tuberculosis, and one idea is that Jewish populations in the past were often confined to urban areas high in Mycobacterium tuberculosis bacteria, and were consequently placed under selective pressures to evolve resistance to the disease.
This high prevalence of Tay-Sachs disease, and other genetic diseases, in some Jewish communities has lead to the establishment of the Committee for Prevention of Genetic Diseases to develop routine genetic testing of young Jewish people worldwide. This was started in the 1980s by Rabbi Joseph Ekstein, who lost four of his five children to the disease. People tested are given a telephone number and a PIN, but are not told their results. Then, when a shidduch (introduction of marriageable singles) is suggested, the candidates can phone the organisation, enter both their PINs, and find out whether their union could result in severely disabled children. Although sometimes receiving criticism, such practices have led to a sharp decline in the occurrence of Tay-Sachs disease in Jewish communities.

Lesch-Nyhan Syndrome
X-linked, 1: 380,000 births
Purines and pyrimidines are the bases found in the nucleotides making up DNA and RNA; Adenine and Guanine are purines whereas Thymine and Cytosine are pyrimidines. These are broken down in different pathways; pyrimidines into beta-amino acids and ammonia, while purines are degraded into urate which is further processed and excreted as uric acid. Lesch-Nyhan syndrome results from inheriting a mutation in a gene involved in purine metabolism leading to increased levels of uric acid. This results in arthritis and severe neurological problems including mental retardation, and self-mutilating behaviour involving biting of the lips, tongue and fingers. This disease was first described in 1964 by Dr. Michael Lesch, a medical student, and Dr. William Nyhan, a paediatrician, at Johns Hopkins Hospital, Baltimore, when they identified symptoms in two affected brothers aged four and eight.
Gout
Gout can result from high concentrations of urate in the blood leading to uric acid crystals forming inside joints, often in the big toe, producing an intense pain; the word gout possibly derives from the Greek “podagra” (Gr. pod; foot, and agra; trap).
Although most cases of gout appear to be the product of diet, mutations in certain genes involved in purine (a component of DNA) metabolism may lead to inherited predispositions to gout. For example, one such gene coding for an enzyme degrading uric acid, when mutated, can lead to an increased predisposition for gout and renal failure. This may explain why certain populations, such as people of the Pacific Islands, and the Maoris of New Zealand, show higher occurrence of this ailment. Incidences of gout in 3,000 year old skeletons on some Pacific islands point to the possibility of a founder effect; the genes responsible for this condition may have been carried to the islands by the early settlers.
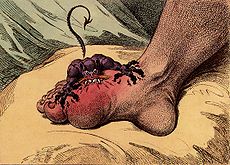

One possible case of familial gout was Charles V of Spain, one of the most powerful rulers of all time during his reign from 1516-1556. Charles V’s gout first became evident when he was 28. As he grew older the attacks of gout increased in frequency and severity. Towards the end he had to be carried around in a specially designed chair as he was barely able to walk or use his hands. Recent analyses of his mummified body revealed high levels of urate deposits confirming his gout. His condition must have influenced many of his decisions ultimately affecting the destiny of many European countries. Charles passed on his vast empire, and his ailment, to his son Phillip. Although, also suffering severely from gout, Phillip supposedly rarely complained about his condition which he regarded as punishment from God for “not being diligent enough to eradicate the protestant heresy”.