
“Blood is inherited and virtue is acquired.”
-Venezuelan Proverb
Mammalian red blood cells (erythrocytes) have a flattened shape with a depressed centre (i.e. biconcave), to optimize exchange of oxygen, and are also flexible so as to fit through the tiny capillaries in the body. In fact the average RBC travels around 1,000 miles through the maze of the body’s circulatory system during their 120 days lifespan before being broken down in the spleen. Defects in the shape of these cells (sickle cell anaemia, hereditary spherocytosis) can lead to anaemia (Gr. a; absence, haem; blood).
These cells are able to carry oxygen due the presence of haemoglobin which is a complex molecule composed of globin proteins, and haem groups whose iron molecules temporarily link to oxygen. Mutations in the production of haem lead to a group of diseases called porphyria, while defects in globin cause thalassaemia. Blood cells are dependent on blood glucose as a source of energy through a biochemical pathway called glycolysis, disruptions in which lead to a disorder known as Glucose-6-phosphate dehydrogenase deficiency – the most commonly inherited enzyme disorder.
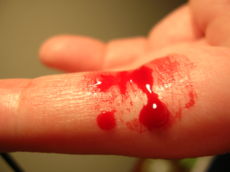
Blood clotting, such as from a cut on the skin, is accomplished by a complicated process involving many different proteins called clotting factors and platelet cells in the blood. Inherited defects in these clotting factors (haemophilia), platelets (thrombocytopenia, Gaucher’s disease, Glanzmann’s thrombasthenia, von Willebrand’s disease) predispose to prolonged bleeding times. In contrast, thrombotic diseases are characterised by increased clotting that can obstruct blood flow.
Sickle Cell Anaemia
Sickle cell anaemia is caused by a specific mutation in a gene encoding one of the β-globin chains making up haemoglobin protein complex. The disorder was first clinically described in 1910 by Chicago doctors James B. Herrick and Ernest Edward Irons who found “peculiar elongated and sickle shaped” cells in the blood of Walter Clement Noel, a 20 year old dental student suffering from anaemia. Although he was readmitted to hospital several times during his degree, Noel completed his studies returning to Grenada to practice dentistry.
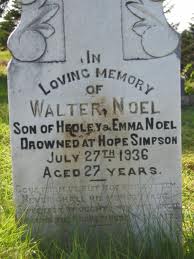

One high profile sufferer is Tionne Tenese “T-Boz” Watkins, the lead singer of the group TLC. She has been in and out of hospital intensive care units, since she was 7 years old with one of her longest hospital stays being for four months in 2002. “Sickle cell is very excruciating pain,” she says. “It feels like somebody is stabbing you over and over again”. Named one of People magazine’s 50 Most Beautiful People in 2000, she is one of the spokespeople for the Sickle Cell Disease Association of America. In the UK and the US all babies are screened for sickle cell anaemia in addition to phenylketonuria and a number of other disorders of metabolism, within the first 2 weeks of birth as part of the standard “heel-prick test” used to collect drops of blood for DNA.
Paul Williams (top right) from The Temptations, also suffered from this disease which drove him to alcohol that wore particularly heavily on his health; a fact not forgotten by the rest of his group who would routinely try to hide alcohol from him. Eventually he had to give up performing with the group he had helped found and in 1974, at the age of 34, was found dead next to his car with an, apparently self-inflicted, gunshot wound to the head.

Hereditary Spherocytosis

Thalassemia
Unlike most genes, there are actually two genes for alpha-globin on each chromosome: the more of these genes that are mutated, the more severe the disease. Defects in all four genes (two on each chromosome) results in early death. Three affected genes leads to the development of anaemia in childhood. Two affected genes can cause a mild anaemia, and if only one of the four alpha genes are affected then no symptoms are usually seen. There is only one gene for beta-globin and if an individual inherits mutations in both parental copies then a severe anaemia called beta-thalassemia major results which, if left untreated, results in death before the age of twenty. If an individual inherits only one mutant beta-globin gene from a parent, beta-thalassemia minor results. This is a mild anaemia which may show some symptoms of fatigue, but in most cases is asymptomatic with many people unaware they have this disorder.
The name thalassemia translates from Greek as “anaemia by-the-sea” (Gr. thalassa; sea) because at one time it was believed that the disease affected only people of Italian or Greek descent. This is now known not to be true as it is also common in Africa, the Middle East and Southeast Asia. The reason it is found in these regions is because, as in the case of sickle cell anaemia, carriers of the disease have a degree of protection against malaria.


Glucose-6-Phosphate Dehydrogenase Deficiency
Red blood cells are rely on glucose as a source of energy through a biochemical pathway using the enzyme Glucose-6-phosphate dehydrogenase (G6PD) amoung others. Deficiency of this enzyme is the most common inherited enzyme deficiency disease in the world and results in an anaemic response to certain foods and chemicals such fava beans (broad beans) and the antimalarial drug, primaquine. This is because G6PD also plays a role in protecting red blood cells against oxidative damage – fava beans contain extremely high amounts of oxidants which can damage G6PD-deficient cells.
G6PD deficiency is possibly the first example of a pharmacogenetic disorder to be described. Pythagoras, in about 500 BC in southern Italy, recognized that some individuals who ate the fava bean became ill, suffering from symptoms of anaemia including jaundice and fatigue, while others could enjoy them with no adverse effects. This leads to the commonly used term “favism” for this disease and Pythagoras used to call on his disciples to abstain from beans. This has however also been interpreted as Pythagoras exhorting his followers to stay away from politics; in ancient Greece and Rome, beans were used as a method of casting votes. Alternatively he may have observed that the high sugar content of beans leads to flatulence! By the late 1940s it was realised that the British, in contrast to Mediterranean populations, rarely developed haemolytic anaemia (an increased destruction of blood cells) on ingestion of fava beans, suggesting a genetic cause. This became even more evident during the Korean War when around 10% of male American soldiers of African or Mediterranean descent developed haemolytic anaemia in response to the antimalarial drug, primaquine, which was later discovered to be also due to G6PD deficiency. This was dramatized in an episode of “M*A*S*H*” in which Cpl. Maxwell Klinger exhibited all the symptoms of malaria but did not have the parasite. However, when he stopped taking primaquine, he recovered completely.
The reason G6PD deficiency is particularly prevalent in people of Mediterranean descent – affecting as many as 1 in 10 – appears again to be due to the protective effect this condition has against malaria. The malaria-causing parasite, plasmodium, requires G6PD for its survival and replication in RBCs. It may be that G6PD-deficient RBCs infected with the Plasmodium, are cleared more rapidly by the spleen, or else that high numbers of oxidants in the blood of G6PD deficient individuals are lethal to Plasmodium. Since the mortality rate for favism is low and the mortality rate for malaria is high, the protective effect of the enzyme deficiency is significant. There are some villages in Sardinia where up to 70% of males are found to be G6PD deficient, possibly reflecting the high infant mortality rates from malaria in these areas after World War II, following deliberate flooding of some coastal areas by departing German troops. G6PD deficiency has also been connected to long life spans (longevity); in centenarians the incidence of G6PD deficiency is, on average, double compared to control groups. Some epidemiological studies have even suggested that G6PD may decrease susceptibility to cancer, cardiovascular disease and stroke.
Porphyria
Haemoglobin is composed of globin proteins combined with a molecule called haem. This consists of an organic ring structure called a porphyrin and an iron atom which is able to bind oxygen. In addition to its role in haemoglobin, haem also has other functions serving as a component of several liver enzymes.
There are 8 steps involved in the making of haem from the 2 molecules glycine and succinyl-CoA, and each step is performed by a specific enzyme encoded by a different gene. If one of the different haem precursor molecules (known as porphyrins) is not modified, it cannot proceed to the next step, instead accumulating in various tissues. The diseases caused by partial defects in these enzymes (a complete absence of haem production is not compatible with life) are known as porphyrias (Gr. porphyros; purple), deriving from purple/red urine colour when the porphyrins are excreted. Affecting around 1 in 20,000 individuals, these porphyrins can accumulate in other parts of the body resulting in different physical and neurological symptoms, dependent upon which enzyme is affected and which porphyrin accumulates as a result.
Acute intermittent porphyria (AIP) results in the accumulation of the porphyrins delta amino levulinic acid (ALA) and porphobilinogen, both of these are neurotoxins. Symptoms include abdominal pain, tremors, seizures and also psychoses, including agitation and hallucinations, which can last for weeks and then quickly abate. Though most people who inherit this trait never develop the symptoms, precipitating factors for such attacks include hormones, drugs and alcohol which induce the production of haem in the liver. It has been suggested that Vincent van Gogh might have suffered from attacks of AIP with several bouts of severe psychosis in his adult life provoking suicide attempts and self-mutilation and necessitating institutionalisation. Factors triggering his symptoms could have included overwork, fasting, malnutrition, and of course his famous absinthe binges, explaining why, during hospitalization, these symptoms generally abated with improved diet and lack of alcohol.
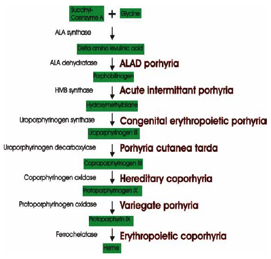

Defects in the forth step of the pathway lead to congenital erythropoietic porphyria (CEP), also known as Gunther’s disease. Here, the porphyin, hydroxymethylbilane, builds up causing severe skin blistering and photosensitivity as the porphyrin absorbs light causing a burning sensation. Teeth can be reddish/brown or even fluorescent due to the porphyrin collecting in the enamel. Many have suggested that cases of CEP in the past could have formed the basis of vampire legends. Symptoms can include: photosensitivity of eyes and skin leading to patients avoiding daylight, severe blistering leading to scaring of the face, teeth enamel stained a red colour, stretching of the lips causing teeth to become more prominent, hypertrichosis leading to a lanugo-like hair growth over the face, and muscle weakness especially in the wrists and fingers. Interestingly, garlic stimulates haem production and so can turn a mild case of porphyria into a very painful one. It has even been suggested that people suffering from porphyria in past centuries might have been prescribed blood to drink in the mistaken belief that this could reduce the anaemia. Furthermore, although porphyrias are rare, it is believed to have been endemic among the Eastern European aristocracy, which was characterised by a high incidence of intermarriage. It is also recorded that Bram Stoker, before writing “Dracula”, met apsychiatric patient who exhibited many of the symptoms (including photosensitivity) associated with CEP, and this could have helped to inspire his novel.
Variegata porphyria (VP) is caused by mutations in the gene for another enzyme in the haem production pathway. The term variegate describes the variable nature of the symptoms; most people do not experience any symptoms while others show a similar condition to that seen in AIP, with abdominal pains, skin rashes, and psychosis in addition to some photosensitivity. King George III, often called the mad king, is suspected of suffering from VP. Between 1788 and 1804, he suffered bouts of ‘‘madness’’ involving a rapid pulse, fever, abdominal pains, constipation, cramp, skin blistering, “port-wine-coloured” urine, and rambling speech degenerating into obscenities and hallucinations. Just as each episode was acute in onset, the recovery phase was equally sudden and rapid. His disease can be traced back to his ancestors, Mary Queen of Scots in the 1500s, and her son, King James I, whose physician noted urine ‘‘red like Alicante wine’’. Other notable royal sufferers are said to include James I’s son Henry, Prince of Wales who had a similar illness and died suddenly, with allegations that he had been poisoned, the Duchess of Orleans who died of a sudden episode of excruciating abdominal pain, Queen Anne, George IV, and Princess Charlotte who died suddenly after childbirth. Prince William of Gloucester (the current duke’s elder brother, who was killed in a plane crash in 1972) had been diagnosed with porphyria suggesting the presence of a gene defect in the family and though it is possible that Queen Victoria may have also carried the gene, no symptoms were reported.


Erythropoietic protoporphyria (Gr. protos; first), which is due to a reduced levels of an enzyme involved in the last step of the pathway converting protoporphyrin IX into haem, leads to very severe photosensitivity, with affected infants screaming soon after being taken out into the sun. It has been suggested that a builder involved in the construction of the Paris Opera house, who lived in the cellars to possibly to avoid sunlight, may have suffered from this disorder inspiring the French writer Gaston Leroux to create “The Phantom of the Opera”.
Hereditary Polycythemia
Some mutations that can result in increased red blood cell numbers. One such mutation increases the efficiency of a hormone secreted from the kidneys, called erythropoietin (EPO), to stimulate the bone marrow to produce more red blood cells. The increased number of these cells and the corresponding increase in oxygen levels in the blood can lead to greater athleticism and endurance. However, this disorder and other disorders leading to an increased number of red blood cells (known as polycythemia) can also lead to capillaries becoming clogged by the increased viscosity of the blood. As a result, untreated sufferers are at a risk of thrombosis, heart attacks and strokes.
The cross-country skier Eero Antero Mäntyranta, one of the most successful skiers Finland has ever produced has hereditary polycythemia following a DNA study done on over 200 members of his family.
As a treatment for anaemia medical scientists have designed drugs that mimic this erythropoietin hormone in order to stimulate red blood cells production. It was not long before some sportsmen realised that EPO drugs could help their blood to carry more oxygen and so allow their bodies to work harder for longer. Consequently, by the late 1990s, EPO-abuse had rocketed with athletes seemingly blind to the dangers associated with increased blood hyperviscosity; it has been linked, for example, to the deaths of 18 Dutch and Belgian cyclists between 1987 and 1990. Despite this, allegedly, EPO abuse is still rife in professional cycling.
Fanconi Anaemia
Red blood cells are produced from stem cells in the red bone marrow of large bones. Diseases resulting in a disruption of this bone marrow can lead to anaemia. One example is Fanconi anaemia, named after the Swiss paediatrician Guido Fanconi, which is due to an altered bone marrow growth predisposing individuals to myeloid leukemia. Failure of the bone marrow to produce blood cells – RBCs, white blood cells and platelets – in this disorder results in a susceptibility to infection, fatigue and haemorrhaging. There are several different types of Fanconi anaemia caused by mutations in different genes involved in the repair of DNA damage and therefore the protection of the body against cancer.
Parents of a girl called Molly Nash, born with Fanconi anaemia, underwent a controversial procedure to conceive another child who could help cure their daughter. They firstly underwent in vitro (Lat. vitro; glass, referring to experiments conducted outside of the body) fertilization, known as IVF, a procedure where egg cells are fertilised outside the woman’s body and grown to the 6 or 8 cell stage and then transferred back to the mother. The embryos were genetically tested for signs of the FA causing gene and only an embryo which did not carry the genetic abnormality, and therefore a safe donor match for Molly, was then implanted into her mother. She subsequently gave birth to a healthy boy named Adam and blood from his umbilical cord containing stem cells was infused into his sister. Both children are fine, and Molly is now able to lead a normal healthy life.
Haemophilia
Deficiencies in some blood clotting factors lead to a bleeding disorder known as haemophilia individuals bleeding for prolonged intervals lasting for days and even weeks in response to only relatively minor injuries or develop internal bleeding. This disorder mostly affects males as the genes for the two major affected blood clotting factors, factor VIII (resulting in haemophilia A) and factor IX (haemophilia B), are located on the X chromosome.
Deficiencies in some blood clotting factors lead to a bleeding disorder known as haemophilia individuals bleeding for prolonged intervals lasting for days and even weeks in response to only relatively minor injuries or develop internal bleeding. This disorder mostly affects males as the genes for the two major affected blood clotting factors, factor VIII (resulting in haemophilia A) and factor IX (haemophilia B), are located on the X chromosome.
First acquiring the name of haemophilia from a Swiss doctor in 1828, this could one of the earliest recognised inherited illnesses. Well over 2,000 years ago, Jewish rabbis referred to a bleeding condition that was fatal to young boys and ran in families, drawing up guidelines to exempt males from circumcision if they had brothers who had previously died from the procedure.
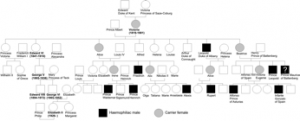
The British royal family has included many males with haemophilia in addition to female carriers including Queen Victoria herself. Recent genetic analysis has shown this to hemophilia B with a mutation in the gene for clotting factor IX. She seems to have spontaneously acquired the haemophilia mutation de nova, as her parents’ families are not known to have had the disease (however, her paternity has been questioned!). And ,in addition to the haemophilia gene, Victoria may have also carried the gene for another blood disorder called porphyria.
She then passed on the mutated clotting factor IX gene to her son Prince Leopold, Duke of Albany and, through several of her daughters, to various royal houses across the continent such as the Spanish, German and Russian royal families. Leopold, finding that his joint pain, which was another symptom of his haemophilia, was aggravated by cold weather, would seek warmer climates abroad during the winter. And it was while in the south of France, in March 1884, that he slipped and fell in the Yacht Club in Cannes, injuring his knee and dying next morning.
His only daughter, Princess Alice of Albany, then passed the gene on to her oldest son, Prince Rupert of Teck, who bled to death after a car accident at the age of 20.
Queen Victoria’s granddaughter, Czarina Alexandra of Russia, passed the disease on to her son Alexei. Desperate to cure their son the Russian royal family turned to the ‘mad monk’ Rasputin whose influence over the family is often cited as a main catalyst for the Russian revolution. His treatment involved hypnosis, a calming confident presence around the boy and family, and exclusion of all the other court physicians who may have been stressful for the young Alexia. It is now known that hypnosis and reduction of stress narrows minor arteries slowing blood circulation and so relieving symptoms. Although it seems unlikely that Rasputin was aware of this, he was certainly not slow to capitalise on his good fortune by spreading his influence over the royal household.



Only a few years after Alexei was born, the Swedish artist Ivar Arosenius, renowned for his paintings of fairy tales died of the disease at a relatively young age. He depicted his affliction in one of his more famous paintings of “Saint George Slaying a Dragon” which can be seen to bleed profusely. The Swedish Haemophilia Society has since established an Arosenius Fund to support haemophilia patients.
Even though the illness has affected those at the highest levels of society, it had remained incurable, with an expected lifespan for sufferers of only 20 years. It is only relatively recently, following the development of blood transfusion, that treatment involving the infusion of blood clotting factors has been available. However, in the UK, contaminated blood has led to widespread infection of HIV and Hepatitis C with around 1,200 people being infected with HIV and more than 4,800 people infected with Hepatitis C from transfusions. From 1986, heat and chemical treatment of blood products has been used to eliminate such viruses, and since 1992 ‘recombinant’ clotting factor, produced by genetic engineering. Unfortunately, there is still no permanent way of replacing or enabling the body to synthesise a functional clotting factor.

Von Willebrand's Disease
Von Willebrand factor (vWF) protein is important in controlling platelet activation. Mutations in the gene coding for this lead to the most common hereditary coagulation disorder, von Willebrand’s disease. Three different types of von Willebrand’s disease exist differing in severity, from the rare severe form to the relatively common mild forms characterised by only slightly prolonged bleeding, possibly affecting as many as 1% of the population. These mild forms can be a complication in child abuse cases, because it causes children to bruise easily.
One person with the severe type of von Willebrand disease is the musician Bill Kozlowski, who was founder of the Alaskan rock band Peabody’s Monster. Much of Kozlowski’s song-writing focuses on living life to the fullest, as he knew his lifespan would be limited.

Thrombasthenia
When blood vessels are damaged, collagen becomes exposed, causing platelet cells to aggregate through binding via a receptor found on their cell surface. Mutations in the gene for this receptor can lead to reduced platelet adhesion and prolonged bleeding times, a condition known as Glanzmann’s thrombasthenia. Another very common mutation in a gene for the receptor, carried by about 20% of the population, can predispose individuals to a higher risk of developing acute coronary artery events.
The sudden death of the Russian Olympic Gold medallist skater Sergei Grinkov has been linked to this phenomenon. On November 20th 1995, at the age of 28, Grinkov died suddenly of a massive heart attack, while in the middle of a practice session. Grinkov’s father also died at the age of 52 of a heart attack, and like his son had none of the risk factors associated with heart disease such as smoking, diabetes, high blood pressure and elevated cholesterol levels. Yet an autopsy revealed that he had severe coronary artery disease and subsequent DNA tests showed that he carried the gene variation.

Thrombotic Diseases
Thrombotic diseases are characterized by the formation of a thrombus, or blood clot, which can obstruct blood flow.
It is suggested that, of all the patients suffering from thromboembolisms, 25-50 percent have a genetic predisposition. This results from defects in genes producing proteins inhibiting blood clot formation as part of the feedback pathway shutting off blood coagulation once a clot is already formed.
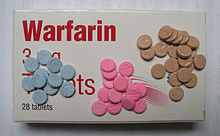
One anticoagulant prescribed to people with increased tendencies for thrombosis is warfarin, which inhibits the production of some of these clotting factors. Initially it was developed as a rat poison, but it was not until a man in the US Navy tried, unsuccessfully, to commit suicide in 1951 by drinking it, that its use as a therapeutic anticoagulant was considered. Interestingly, warfarin was used to treat one world leader, Dwight Eisenhower after his heart attack in 1955, and possibly used to dispatch another, Joseph Stalin in 1953!